
3.Симптомы и диагностика
Синдром Марфана обнаруживается уже в раннем возрасте; он может прогрессировать или оставаться относительно стабильным. Классической триадой считают деформацию опорно-двигательного аппарата, офтальмологическую патологию и сердечнососудистую недостаточность.
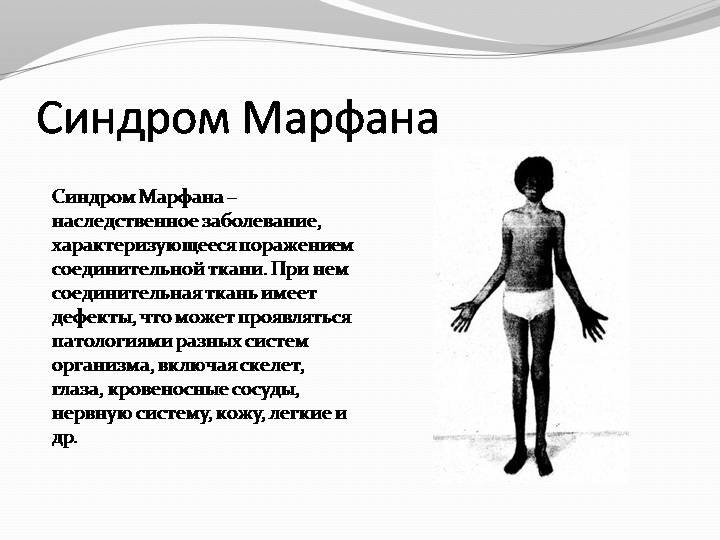
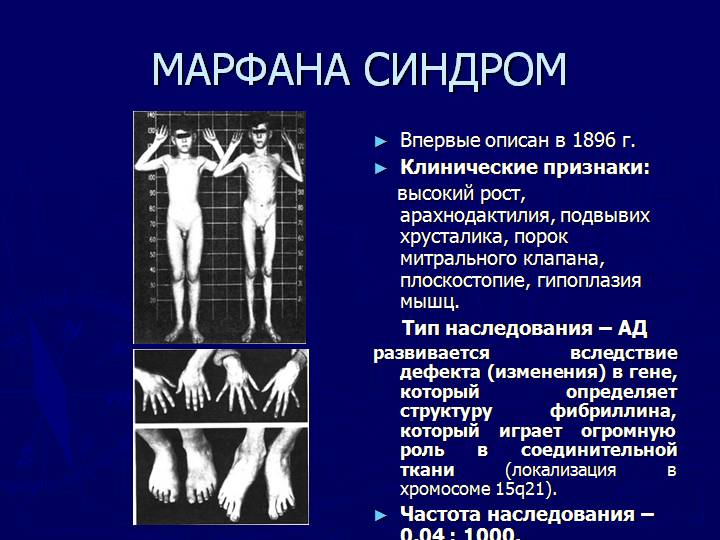
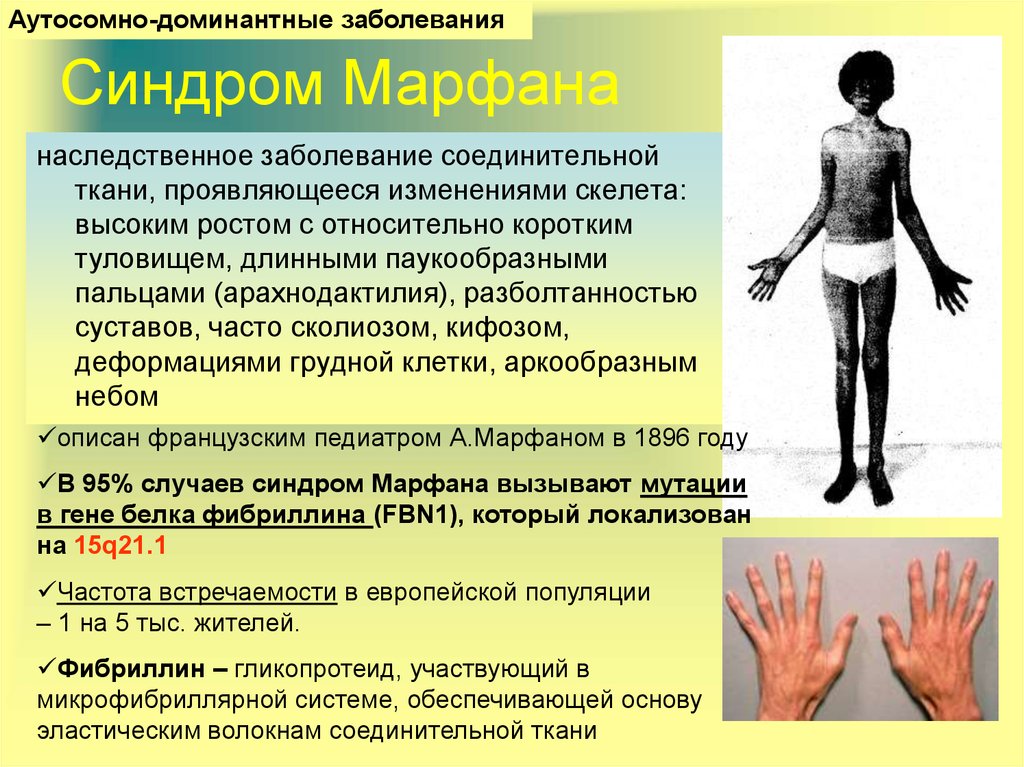
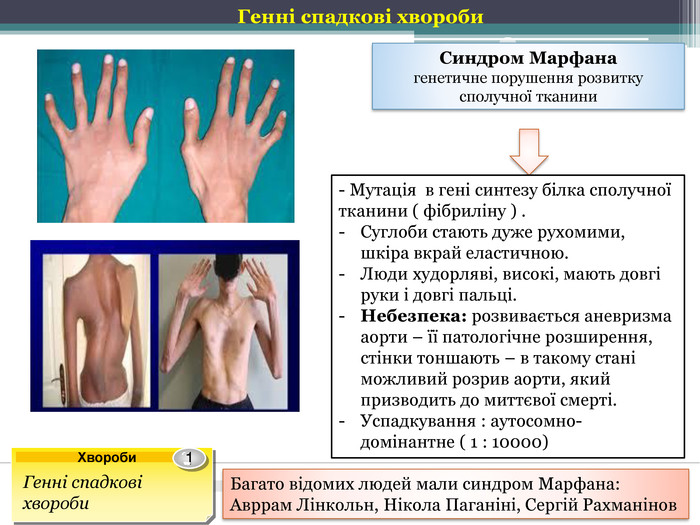
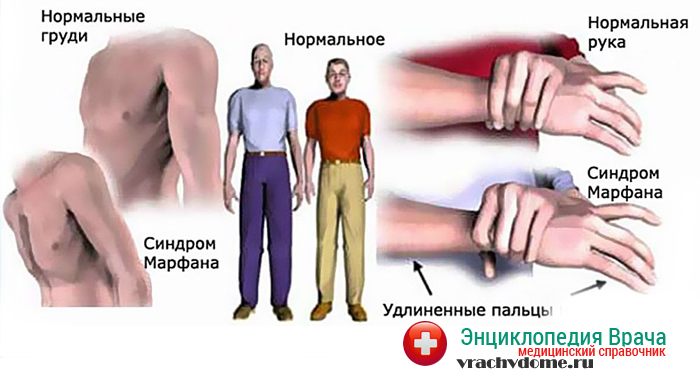
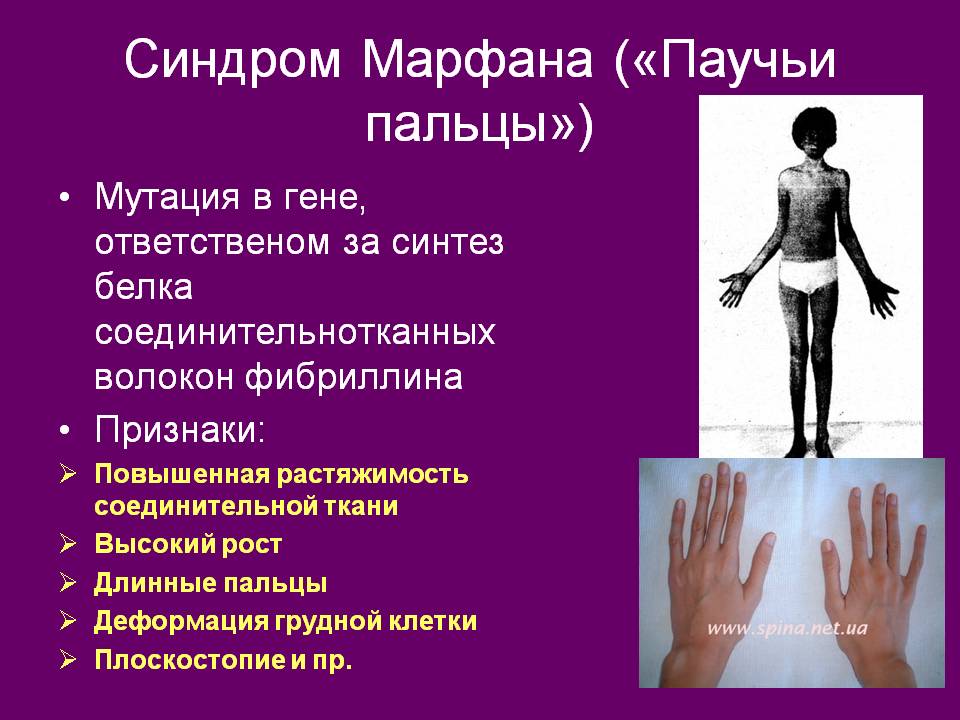
Больные отличаются высоким ростом и диспластичным телосложением: непропорционально длинные конечности, «паучьи пальцы» (арахнодактилия), килевидная или вдавленная грудная клетка, сложные искривления позвоночника (лордоз, кифосколиоз), вальгусная или варусная конфигурация ног, патологическая подвижность суставов, плоскостопие и т.д.
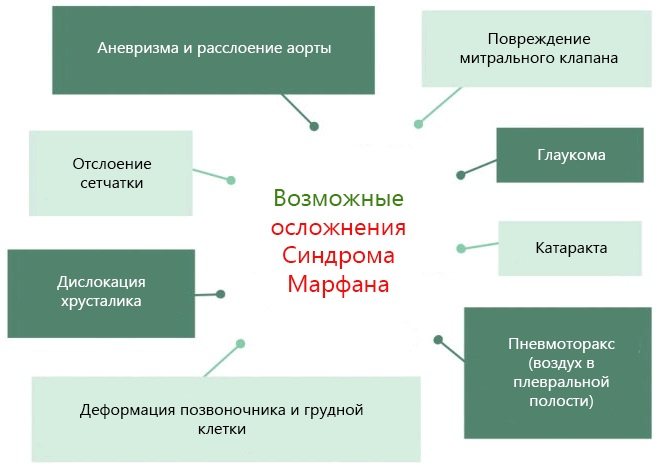
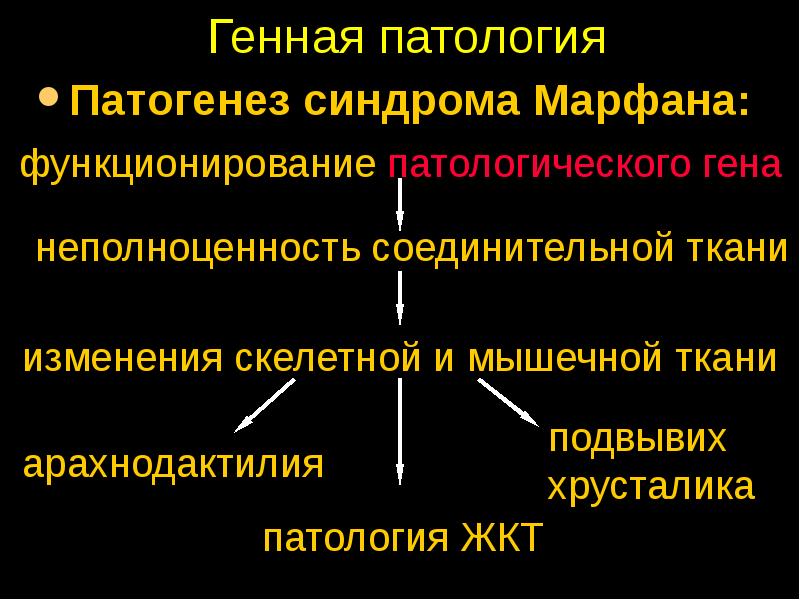
Характерные диспропорции обнаруживаются также в строении челюстно-лицевых структур. Явно недостаточно развиты мышечная и жировая ткани. Типичным признаком является аневризма аорты и/или других крупных сосудов, в т.ч. легочных и мозговых; встречается множество различных пороков сердца. Столь же полиморфными и тяжелыми могут быть аномалии развития органов дыхания, пищеварения, а также центральной нервной и зрительной систем (в частности, офтальмологически выявляется выраженная миопия или гиперметропия, подвывих хрусталика, полное отсутствие радужки или хрусталика, удлинение глазного яблока и т.п.).
Комплекс столь критичных по сути аномалий может быть выражен относительно мягко, однако в целом синдром Марфана обычно нуждается в квалифицированном терапевтическом наблюдении и сопровождении, – в противном случае и качество, и продолжительность жизни резко ограничиваются (в отсутствие курации непосредственной причиной смерти становится, как правило, хроническая или острая, наступающая вследствие разрыва аневризмы, сердечная недостаточность).
Диагноз устанавливается путем изучения семейного анамнеза и всестороннего обследования: практически любой из существующих сегодня диагностических методов выявляет те или иные характерные изменения.
Методы диагностики синдрома Марфана
Синдром Марфана диагностируют на основании клинических проявлений, генетического исследования, а также данных эхокардиографии или магнитно-резонансной томографии. С помощью эхокардиографии и МРТ выявляют патологические изменения в области аортального корня и клапанов. Аномальные изменения в хрусталике видны при использовании щелевой лампы, а расширение дурального мешка спинного мозга показывает МРТ. При осмотре глаз окулист видит типичный признак генетической аномалии в виде подвывиха хрусталика.
Тем не менее, в ряде случаев постановка корректного диагноза вызывает трудности, так как многие пациенты не обладают яркими признаками синдрома Марфана. Также у них могут отсутствовать изменения со стороны биохимии и гистологии. В данном случае клиницистам следует ориентироваться на сбор анамнеза, объективный осмотр и генетическое исследование.
Диагностика синдрома Марфана
В диагностическую программу синдрома Марфана входит:
- сбор жалоб и анамнеза больного (ключевым моментом является составление генеалогического древа, по которому можно проследить аутосомно-доминантный тип наследования заболевания);
- осмотр и физикальное обследования ребенка: наличие характерной деформации скелета и гипермобильности суставов, аускультативной картины порока сердца и нарушение зрения являются клиническими критериями данного заболевания;
- фенотипические диагностические тесты: соотношение длины кисти к росту ребенка, индекс телосложения Варги, тест на арахнодактилию и другие;
- электрокардиография (ЭКГ) и эхокардиография (ЭхоКГ) используются с целью идентификации патологии сердца;
- аортография проводится с целью измерения диаметра сосуда и выбора дальнейшей тактики лечения (консервативное или хирургическое);
- офтальмологическое обследование верифицирует характерные нарушения строения глаз;
- рентгенологическое исследование органов грудной клетки (выявляет уменьшение размеров легких, изменение конфигурации сердца, расширение дуги аорты);
- рентгенография тазобедренных суставов (определение протрузии вертлужной впадины);
- рентгенография позвоночника (визуализация деформации позвоночника);
- КТ и МРТ используются для диагностики дилатации или аневризмы аорты;
- лабораторное исследование мочи на экскрецию гликозаминогликанов;
- ДНК-анализ для определения мутации гена FBN1.
Симптомы
Фенотип больных характеризуется определённой протяжённостью: начиная от лёгких, «мягких» форм соединительнотканной дисплазии, встречающихся и в общей популяции — до случаев с угрожающими жизни системными расстройствами.
Органы зрения: у половины больных диагностируется подвывих хрусталика; у лиц с выраженной миопией повышен риск отслойки сетчатки.
Мышечно-скелетная система: арахнодактилия, долихостеномелия, деформации позвоночника (сколиоз, лордоз, гиперкифоз), деформация передней стенки грудной клетки (вдавленная грудь, «куриная грудь»), гипермобильность суставов, плоская стопа, высокое готическое нёбо, недоразвитие вертлужной впадины, врождённые контрактуры локтей и пальцев, мышечная гипотония.
Сердечно-сосудистая система: пролапс митрального клапана отмечается в 80 % случаев; со временем створки клапанов утолщаются, становясь гистологически миксоматозными; дилатация корня аорты начинается с синуса Вальсальвы и прогрессирует с возрастом (у женщин отмечается более медленное прогрессирование) и в конечном итоге может приводить к расслаивающейся аневризме аорты.
Другие системы органов: у 5 % больных отмечаются спонтанные пневмотораксы; характерны стрии на коже (striae atrophicae) в областях плеч, груди, поясницы; у большинства больных наблюдается сужение нервного канала в пояснично-крестцовом отделе; нередко диагностируются кистозные образования в печени и почках, которые увеличиваются с возрастом и обычно клинически не значимы.
Самые частые симптомы расстройства
Типичные симптомы для синдрома Ретта – мышечные и двигательные нарушения. Мышцы находятся в гипертонусе или же, наоборот, теряют его. В этом случае у ребенка развивается неправильное положение тела, прогрессирует частичные параличи и нарушение координации. Например, девочки скрещивают ноги во время ходьбы.
Синкинезии – патологические сокращения мышц, возникают вслед за произвольным движением: простая улыбка способна вызвать резкий взмах ногой. Такое явление постепенно приводит к повреждению суставов, сухожилий и связок, провоцирует ортопедические нарушения. Последние проявляются во всевозможных деформациях и также очень часто сопровождают таких детей. Среди них выделяют вывих тазобедренного сустава, провоцируемый малой подвижностью.
Статическая деформация стопы чаще развивается из-за нарушенного мышечного тонуса. Распространенной считается патология под названием «конская стопа», связанная со снижением подвижности голеностопного сустава. Ее можно узнать по пятке, которая не достигает земли, стопа при этом смещается кнаружи или вовнутрь. Причина патологии – гипертонус икроножной мышцы.










Сколиоз – боковое искривление позвоночника, который провоцирует массу проблем у таких пациентов: деформации суставов и костей, боли во время ходьбы, в стоячем или сидячем положении, утрата способности передвигаться. Сколиоз грудного отдела вызывает легочную недостаточность. Появляются также проблемы с пищеварением.
У детей с синдромом Ретта наблюдается повышенное слюнотечение. Но это происходит не из-за избытка количества слюны, а потери способности сглатывать ее.
Нарушение питания может развиваться из-за частых приступов тошноты. Она появляется на любые аспекты питания: на определенный продукт, его температуру, на способ приготовления. Так, ребенок способен отрицательно реагировать на пищу, поданную кусочками, или на комочки в блюде.
Постоянная тошнота провоцирует отказ от питания, а значит, потерю в весе.
Плохое сглатывание слюны, которая регулирует кислотность в желудке, и повышенное внутрибрюшное давление вызывают желудочно-пищеводный рефлюкс, то есть забрасывание содержимого желудка в пищевод. Это чревато такими последствиями, как воспаление стенки пищевода, респираторные инфекции.
Малоподвижный образ жизни, неврологические расстройства, неправильное питание провоцируют возникновение запоров у детей с синдромом Ретта. Они носят тяжелый характер, поскольку способны вызывать закупорку кишечника и сильные боли.
Повышенное слюнотечение, тошнота, рефлюкс снижают потребление ребенком пищи и даже развивают на нее негативную реакцию. В результате этого ребенок теряет в весе. Этот процесс стоит строго контролировать, поскольку он чреват истощением.
Другое тяжелое расстройство связано с работой дыхательной системы, развивающееся вплоть до приступов апноэ. Это явление настолько часто среди детей с синдромом, что нередко стает причиной их гибели.
Важными патогномоничными признаками синдрома считаются проявления аутизма. Именно из-за них заболевание изначально считали одной из форм этого расстройства, а в настоящее время относят к болезням аутистического спектра.
Аутистические признаки проявляются в отстранении от окружающего мира, в том числе и от родственников. Ребенок замыкается в себе, может не откликаться, когда его зовут. Предпочитает одиночество. Дети боятся чужих людей и непривычных ситуаций.
Лицо такого ребенка становится похожим на каменное. Взгляд блуждающий или устремлен в одну точку. Поведение часто непредсказуемо: случаются приступы неутомимого смеха или плача. Склонны к самоповреждениям: царапают кожу, кусают пальцы, вырывают волосы.
Методы терапии
Прежде всего, лечение заключается:
- в назначении бета-адреноблокаторов;
- в хирургическом вмешательстве при патологиях клапанов и аорты;
- в хирургической коррекции патологий позвоночника.
Из бета-блокаторов пациентам целесообразно назначать препараты в виде пропранолола или атенолола. Это помогает предотвратить серьёзные осложнения со стороны сосудов и сердца. Бета-блокаторы уменьшают интенсивность сокращения сердечной мышцы, снижая её нагрузку, останавливают процесс расслоения аорты и снижают риск развития аневризмы. Если аневризма достигает критических размеров, пациентам показано хирургическое вмешательство.
В качестве консервативного лечения сколиоза, обычно, применяют фиксацию позвоночника, но если он искривлён от 40 градусов и более, операция является более предпочтительным методом.
Всем пациентам, страдающим синдромом Марфана, нужно каждый год проходить обследование у невролога, кардиолога и окулиста, с генетическим консультированием по показаниям.
Другие заболевания из группы Болезни органов дыхания:
| Агенезия и Аплазия |
| Актиномикоз |
| Альвеококкоз |
| Альвеолярный протеиноз легких |
| Амебиаз |
| Артериальная легочная гипертония |
| Аскаридоз |
| Аспергиллез |
| Бензиновая пневмония |
| Бластомикоз североамериканский |
| Бронхиальная Астма |
| Бронхиальная астма у ребенка |
| Бронхиальные свищи |
| Бронхогенные кисты легкого |
| Бронхоэктатическая болезнь |
| Врожденная долевая эмфизема |
| Гамартома |
| Гидроторакс |
| Гистоплазмоз |
| Гранулематоз вегенера |
| Гуморальные формы иммунологической недостаточности |
| Добавочное легкое |
| Ехинококкоз |
| Идиопатический Гемосидероз легких |
| Идиопатический фиброзирующий альвеолит |
| Инфильтративный туберкулез легких |
| Кавернозный туберкулез легких |
| Кандидоз |
| Кандидоз легких (легочный кандидоз) |
| Кистонозная Гипоплазия |
| Кокцидиоилоз |
| Комбинированные формы иммунологической недостаточности |
| Кониотуберкулез |
| Криптококкоз |
| Ларингит |
| Легочный эозинофильный инфильтрат |
| Лейомиоматоз |
| Муковисцидоз |
| Мукороз |
| Нокардиоз (атипичный актиномикоз) |
| Обратное расположение легких |
| остеопластическая трахеобронхопатия |
| Острая пневмония |
| Острые респираторные заболевания |
| Острый абсцесс и гангрена легких |
| Острый бронхит |
| Острый милиарный туберкулез легких |
| Острый назофарингит (насморк) |
| Острый обструктивный ларингит (круп) |
| Острый тонзиллит (ангина) |
| Очаговый туберкулез легких |
| Парагонимоз |
| Первичный бронхолегочный амилоидоз |
| Первичный туберкулезный комплекс |
| Плевриты |
| Пневмокониозы |
| Пневмосклероз |
| Пневмоцитоз |
| Подострый диссеминированный туберкулез легких |
| поражение газами промышленного происхождения |
| Поражение легких вследствие побочного действия лекарственных препаратов |
| поражение легких при диффузных болезнях соединительной ткани |
| Поражение легких при болезнях крови |
| Поражение легких при гистиоцитозе |
| Поражение легких при дефеците а 1- антитрипсина |
| поражение легких при лимфогранулематозе |
| Поражение легких при синдроме Стивенса-Джононса |
| Поражения легких отравляющими веществами |
| Пороки развития легких |
| Простая Гипоплазия |
| Радиационные поражения легких |
| Рецидивирующий бронхит у детей |
| Саркаидоз органов дыхания |
| Секвестрация легкого |
| Синдром гудпасчера |
| Синдром Маклеода |
| Синдром Мендельсона |
| Синусит |
| Спонтанный пневмоторакс |
| Споротрихоз |
| Стафилококковые деструкции легких у детей |
| Стенозы и трахеи крупных бронхов |
| Стенозы и трахеи крупных бронхов |
| Стрептококковый фарингит |
| Сфеноидальный синусит (сфеноидит) |
| Токсоплазмоз |
| Трахеальный бронх |
| Трахеит |
| Трахеобронхомегалия |
| Тромбоэмболия легочной артерии (ТЭЛА) |
| Туберкулез внутригрудных лимфатических узлов (бронхоадениты) |
| Туберкулез бронхов, трахеи, верхних дыхательных путей |
| Туберкулез гортани |
| Туберкулез легких |
| Туберкулез полости рта, миндалин и языка |
| Туберкулезная интоксикация у детей и подростков |
| Туберкулезный плеврит |
| Туберкулема легких |
| Фарингит |
| Фиброзно-кавернозный туберкулез |
| Фронтит (острый фронтальный синусит) |
| Хроническая пневмония |
| Хроническая пневмония у детей |
| Хронический абсцесс легких |
| Хронический бронхит |
| Хронический гематогенно-диссеминированный туберкулез легких |
| Хроническое легочное сердце |
| Цирротический туберкулез легких |
| Шистосомозы |
| Экзогенный аллергический альвеолит |
| Эмфизема легких |
| Эпиглоттит |
| Этмоидальный синусит (этмоидит) |
Диагностика
УЗИ человека с синдромом Марфана, показывающее расширенный корень аорты
Диагностические критерии MFS были согласованы на международном уровне в 1996 году. Однако синдром Марфана часто трудно диагностировать у детей, поскольку они обычно не проявляют симптомов до достижения полового созревания. Диагноз основывается на семейном анамнезе и комбинации основных и второстепенных показателей расстройства, редко встречающихся в общей популяции, которые встречаются у одного человека, например: четыре скелетных признака с одним или несколькими признаками в другой системе организма, такой как глазная и сердечно-сосудистые у одного человека. Следующие состояния могут быть результатом MFS, но могут также возникать у людей без каких-либо известных основных заболеваний.






- Аневризма или расширение аорты
- Арахнодактилия
- ГЭРБ
- Двустворчатый аортальный клапан
- Кисты
- Кистозный медиальный некроз
- Дегенеративная болезнь диска
- Искривленная перегородка
- Дуральная эктазия
- Ранняя катаракта
- Ранняя глаукома
- Ранний остеоартроз
- Эктопия лентис
- Эмфизема
- Ирис колобома
- Выше среднего роста
- Учащенное сердцебиение
- Грыжи
- Небо с высокой аркой
- Гипермобильность суставов
- Кифоз (сгорбившись)
- Негерметичный сердечный клапан
- Неправильный прикус
- Микрогнатия (малая нижняя челюсть)
- Пролапс митрального клапана
- Миопия (близорукость)
- Обструктивная болезнь легких
- Остеопения (низкая плотность костной ткани)
- Pectus carinatum или экскаватум
- Pes planus ( плоскостопие )
- Пневмоторакс (коллапс легкого)
- Отслойка сетчатки
- Сколиоз
- Апноэ во сне
- Растяжки не от беременности или ожирения
- Зубы скучены
- «Узкое, худое лицо»
- Дисфункция височно-нижнечелюстного сустава (ВНЧС)
Пересмотренная гентская нозология
Знак большого пальца; верхний : нормальный, нижний : синдром Марфана
В 2010 году нозология Гента была пересмотрена, и новые диагностические критерии заменили предыдущее соглашение, заключенное в 1996 году. Семь новых критериев могут привести к постановке диагноза:
При отсутствии семейного анамнеза MFS:
- Z-показатель корня аорты ≥ 2 И эктопия lentis
- Z-показатель корня аорты ≥ 2 И мутация FBN1
- Z-оценка корня аорты ≥ 2 И системная оценка *> 7 баллов
- Ectopia lentis И мутация FBN1 с известной патологией аорты
При наличии семейного анамнеза MFS (как определено выше):
- Эктопия лентис
- Системный балл * ≥ 7
- Z-показатель корня аорты ≥ 2
- Очки за системный балл:
- Знак запястья И большого пальца = 3 (знак запястья ИЛИ большого пальца = 1)
- Деформация Pectus carinatum = 2 (асимметрия грудной клетки или грудной клетки = 1)
- Деформация заднего отдела стопы = 2 (плоская стопа = 1)
- Эктазия твердой мозговой оболочки = 2
- Протрузия вертлужной впадины = 2
- пневмоторакс = 2
- Уменьшение соотношения верхнего и нижнего сегментов И увеличение руки / роста И отсутствие тяжелого сколиоза = 1
- Сколиоз или грудопоясничный кифоз = 1
- Уменьшенное разгибание локтей = 1
- Черты лица (3/5) = 1 ( долихоцефалия , энофтальм , опускание глазных щелей , гипоплазия скуловой кости , ретрогнатия )
- Бороздки кожи ( растяжки ) = 1
- Близорукость > 3 диоптрии = 1
- Пролапс митрального клапана = 1
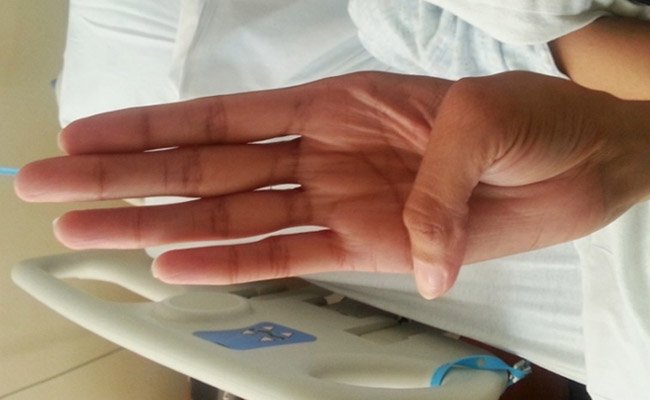
Знак большого пальца (знак Стейнберга) вызывается просьбой человека согнуть большой палец как можно дальше, а затем сомкнуть пальцы над ним. Положительный признак большого пальца – это когда вся дистальная фаланга видна за локтевым краем кисти, что вызвано сочетанием гипермобильности большого пальца, а также большого пальца, который длиннее обычного.
Знак запястья (знак Уокера-Мердока) вызывается, когда человека просят обхватить большим пальцем и пальцами одной руки другое запястье. Положительным признаком запястья является перекрытие мизинца и большого пальца из-за сочетания тонких запястий и длинных пальцев.
Дифференциальная диагностика
Многие другие расстройства могут вызывать те же характеристики тела, что и синдром Марфана. Генетическое тестирование и оценка других признаков и симптомов могут помочь их дифференцировать. Ниже приведены некоторые расстройства, которые могут проявляться как «марфаноид»:
- Врожденная контрактурная арахнодактилия , также известная как синдром Билса-Хехта
- Синдром Элерса-Данлоса
- Гомоцистинурия
- Синдром Лойса-Дитца
- MASS фенотип
- Множественная эндокринная неоплазия 2В типа
- Синдром Шпринцена – Гольдберга
- Синдром Стиклера
Контроль и лечение заболевания
Дети с синдромом Марфана должны постоянно быть под медицинским и домашним наблюдением. Их организм растет и развивается довольно быстро, особенно в подростковый период, и тогда нужны будут осмотры окулиста, ортопеда и других специалистов, проведение при необходимости лечебных мероприятий. Если соблюдать рекомендации для больных синдромом Марфана, то можно сохранить здоровье надолго.
Это не означает, что активность им противопоказана совсем. Такие люди могут и должны поддерживать физическую форму. Например, езда на велосипеде, медленные танцы, пешие прогулки, плавание вполне подходят в данной ситуации.

Многие проявления синдрома Марфана контролируются с помощью лекарств, а при необходимости — хирургическими способами. Так, бета-адреноблокаторы применяют для понижения артериального давления и уменьшения износа стенок сосудов, что тормозит процесс прогрессирования аневризмы аорты. Если же она достигает слишком больших размеров или беспокойство вызывает сердечный клапан, может быть назначена хирургическая операция.
Детям, страдающим миопией или синдромом «ленивого глаза», назначаются коррекционные очки или контактные линзы.
При развитии сколиоза, возможно, придется носить специальный поддерживающий корсет, заниматься ЛФК. В особо тяжелых ситуациях при сильной деформации грудной клетки или позвоночника может потребоваться операция. В некоторых странах дети и подростки с синдромом Марфана носят браслет с информацией. Это понадобится в критическом случае, чтобы врачи знали диагноз пациента.
Синдром Марфана — что это? Тип наследования

Данное заболевание имеет аутосомно-доминантный характер. В его основе лежат мутации в гене FBN1, отвечающем за синтез фибриллина — одного из самых важных белков для нормальной жизнедеятельности, который поддерживает эластичность соединительной ткани. При болезни Марфана возникает его недостаток, а это приводит к тому, что соединительная ткань теряет прочность и упругость, человек не может выдерживать большие физиологические нагрузки. Также страдает аорта и цинновая связка глаза, которая содержит большое количество фибриллина. Эластических волокон много во всем теле человека, но самое большое их сосредоточение именно в этих структурах. Нередко смерть больного происходит именно от аневризмы аорты.
Также было выявлено, что вероятность рождения младенца с синдромом Марфана увеличивается в зависимости от возраста отца: чем он старше, тем больше риск.
Нетипичная картина
Наряду с типичной формой заболевания, описанной выше, встречаются и атипичные формы. Они имеют свои особенности, от которых зависит тяжесть заболевания.




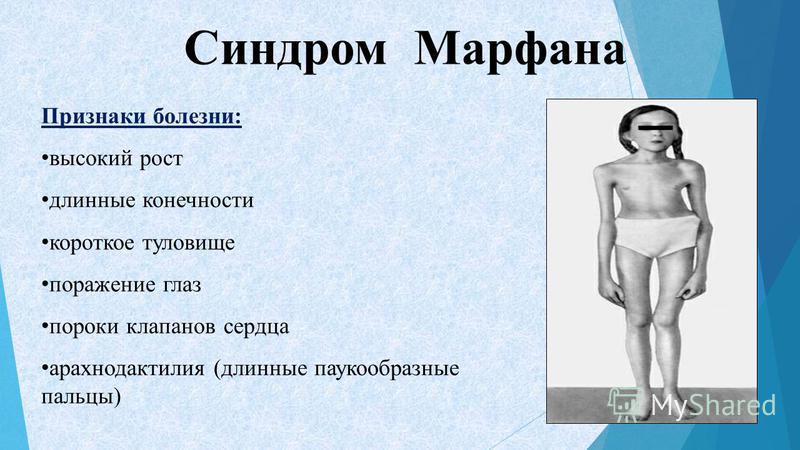

- Zapella – форма синдрома с неярко выраженными признаками. Речь частично сохранена, умеренно выражен сколиоз, умственная отсталость средней степени тяжести. Физически развиваются нормально.
- Hanefeld – в клинической картине преобладает раннее развитие судорожных приступов. Часто они случаются даже до появления умственной деградации.
- Rolando – на первый план выходят признаки задержки психомоторного развития. Ребенок теряет возможность передвигаться, нарастает стереотипия движений, его беспокоят дыхательные нарушения.
Синдром Ретта – сложное генетическое заболевание. Прежде всего, его сопровождает полная умственная деградация и психоневрологические нарушения, влекущие за собой многочисленные патологии других систем организма.
К сожалению, в мире еще не существует способа кардинального искоренения болезни, хотя ученые ведут постоянные разработки в этом направлении.
Лечение синдрома сводится к трем основным направлениям. Медикаментозная терапия назначается для купирования судорожных припадков и стимуляции работы головного мозга.
Диетотерапия включает в себя контроль массы тела, употребление в пищу высококалорийных, витаминизированных продуктов.
Однако наибольшее внимание уделяется реабилитационным мероприятиям, направленным на укрепление опорно-двигательного аппарата и поддержание умственного, психомоторного развития
Важно сохранить комплексный, всесторонний подход к проблеме. Такие дети нуждаются в постоянной поддержке со стороны взрослых и веры в них
Сотрудничество с ними, как с полноценной ячейкой общества, способствует их лучшей адаптации в социуме и более благоприятному развитию
Такие дети нуждаются в постоянной поддержке со стороны взрослых и веры в них. Сотрудничество с ними, как с полноценной ячейкой общества, способствует их лучшей адаптации в социуме и более благоприятному развитию
Важно сохранить комплексный, всесторонний подход к проблеме. Такие дети нуждаются в постоянной поддержке со стороны взрослых и веры в них
Сотрудничество с ними, как с полноценной ячейкой общества, способствует их лучшей адаптации в социуме и более благоприятному развитию.
Другие заболевания из группы Болезни костно-мышечной системы и соединительной ткани:
| Cиндром Шарпа |
| Алкаптонурия и охронотическая артропатия |
| Аллергический (эозинофильный) гранулематозный ангиит (синдром Черджа-Штрауса) |
| Артриты при хронических заболеваниях кишечника (неспецифическом язвенном колите и болезни Крона) |
| Артропатия при гемохроматозе |
| Болезнь Бехтерева (анкилозирующии спондилоартрит) |
| Болезнь Кавасаки (слизистокожножелезистыи синдром) |
| Болезнь Кашина-Бека |
| Болезнь Такаясу |
| Болезнь Уипла |
| Бруцеллезный артрит |
| Внесуставный ревматизм |
| Геморрагический васкулит |
| Геморрагический васкулит (болезнь Шенлейна – Геноха) |
| Гигантоклеточный артериит |
| Гидроксиапатитная артропатия |
| Гипертрофическая легочная остеоартропатия (болезнь Мари – Бамбергера) |
| Гонококковый артрит |
| Гранулематоз Вегенера |
| Дерматомиозит (ПМ) |
| Дерматомиозит (полимиозит) |
| Дисплазия тазобедренного сустава |
| Дисплазия тазобедренных суставов |
| Диффузный (эозинофильный) фасциит |
| Зоб |
| Иерсиниозный артрит |
| Интермиттирующий гидрартроз (перемежающаяся водянка сустава) |
| Инфекционный (пиогенный) артрит |
| Иценко – Кушинга болезнь |
| Лаймовская болезнь |
| Локтевой стилоидит |
| Межпозвонковый остеохондроз и спондилез |
| Миотендинит |
| Множественные дизостозы |
| Множественный ретикулогистиоцитоз |
| Мраморная болезнь |
| Невралгия позвоночного нерва |
| Нейроэндокринная акромегалия |
| Облитерирующий тромбангиит (болезнь Бюргера) |
| Опухоль верхушки легкого |
| Остеоартроз |
| Остеопойкилия |
| Острый инфекционный артрит |
| Палиндромный ревматизм |
| Периартрит |
| Периодическая болезнь |
| Пигментный виллезанодулярный синовит (синовит геморрагический) |
| Пирофосфатная артропатия |
| Плексит плечевого сустава |
| Пневмокониоз |
| Подагра |
| Пояснично-крестцовый плексит |
| Псориатический артрит |
| Реактивный артрит (артропатия) |
| Ревматизм |
| Ревматическая полимиалгия |
| Ревматоидный артрит |
| Рецидивирующий полихондрит |
| Саркоидоз |
| Синдром (болезнь) Рейтера |
| Синдром Барре – Льеу |
| Синдром Бехчета |
| Синдром Гудпасчера |
| Синдром запястного канала |
| Синдром тарзального канала |
| Синдром Титце |
| Синдром Фелти |
| Синдром Шегрена |
| Синдром Элерса – Данло |
| Синовиома |
| Сирингомиелия |
| Системная красная волчанка |
| Системная красная волчанка (СКВ) |
| Системная склеродермия |
| Сифилитический артрит |
| Смешанная криоглобулинемия (криоглобулинемическая пурпура) |
| Смешанное заболевание соединительной ткани |
| Сывороточная и лекарственная болезнь |
| Тендовагинит |
| Туберкулез позвоночника |
| Туберкулезный полиартрит |
| Узелковый полиартериит |
| Фиброзит (фасцииты и апоневрозиты) |
| Хондродисплазия |
| Хондроматоз суставов |
| Шейный плексит |
Примечания
- Marfan A. B. Un cas de deformation congenital des quatre membres plus prononcee aux extremities caracterisee par l’allongement des os avec un certain degre d’amincissement. // Bulletins Et Memoires De La Societe Medicale Des Hopitaux De Paris. — 1896. — Vol. 13. — PP. 220—226.
- Pyeritz R.E. Disorders of fibrillins and microfibrilogenesis: Marfan syndrome, MASS phenotype, contratural arachnoductyly and related conditions. In: Rimoin D.L., Connor J.M., Pyeritz R.E. (eds). «Principles and Practice of Medical Genetics», 3rd ed. New York: Churchill Livingstone, in press 1996.
- Goldman’s Cecil Medicine 978-1-4557-1167-3,
Лечение
Лечение — преимущественно симптоматическое, направлено на облегчение тех или иных проявлений заболевания. Больным необходимо проходить расширенное ежегодное медицинское обследование с обязательным участием офтальмолога, кардиолога и ортопеда.
Большинство клинических исследований поддерживают профилактическое употребление бета-адреноблокаторов с раннего возраста для предотвращения расслаивающейся аневризмы аорты. В случае выраженной дилатации корня аорты проводится его хирургическая коррекция. Показанием для операции у взрослых больных является достижение максимального диаметра корня аорты 50 мм.
Лечение болезни паучего пальца
Терапия синдрома паучьих пальцев проводится комплексно. Основные методы лечения:
- Использование препаратов, регулирующих уровень коллагена. Лекарства способствуют продукции компонента в необходимом количестве и устраняют его нехватку.
- Улучшение вещественного обмена. Врач назначает прием аскорбиновой кислоты в большом количестве и В витамина.
Если речь идет о терапии болезни Марфана, используют антиокседантные препараты, средства на основе стероидов анаболического типа и небольшое количество антиагрегантов. Кроме медикаментозного лечения врач назначает диету, ограничивающую потребление белка животного происхождения.



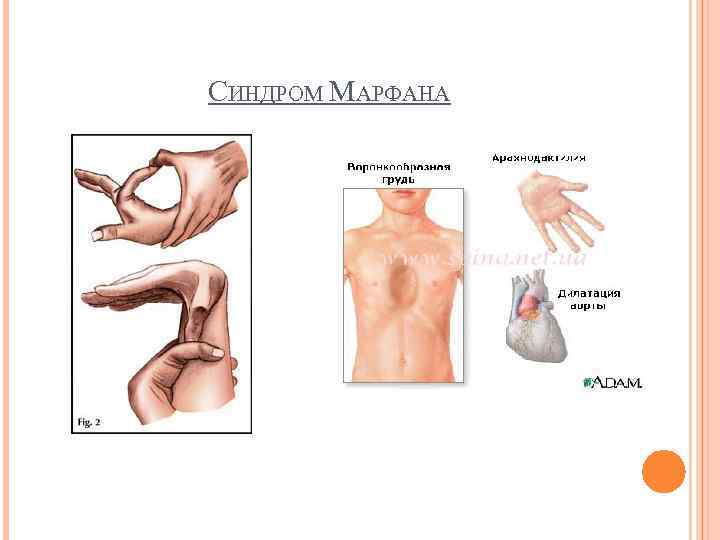



Когда пациент страдает от астигматизма, ему нужно носить очки или линзы. Это позволит глазам не напрягаться и предупредит последующее ухудшение зрения.
При нарушении нормальной формы грудины с образованием воронки показана торакопластика. Задача процедуры – остановить уменьшение области за ребрами во избежание травмирования и сдавливания внутренних систем.
Патологии сердца лечатся с помощью операции. Состояние аорты нормализуют путем пластики, а вместо клапанов устанавливают протезы.
В обязательном порядке показана лечебная гимнастика. Подобрать комплекс упражнений поможет врач. Сначала ЛФК выполняется под наблюдением доктора, затем больной может делать зарядку дома. Физкультура способствует повышению мышечного тонуса и гибкости позвоночника.
Для предупреждения прогрессирования болезни прибегают к отдыху в санаториях и на курортах.

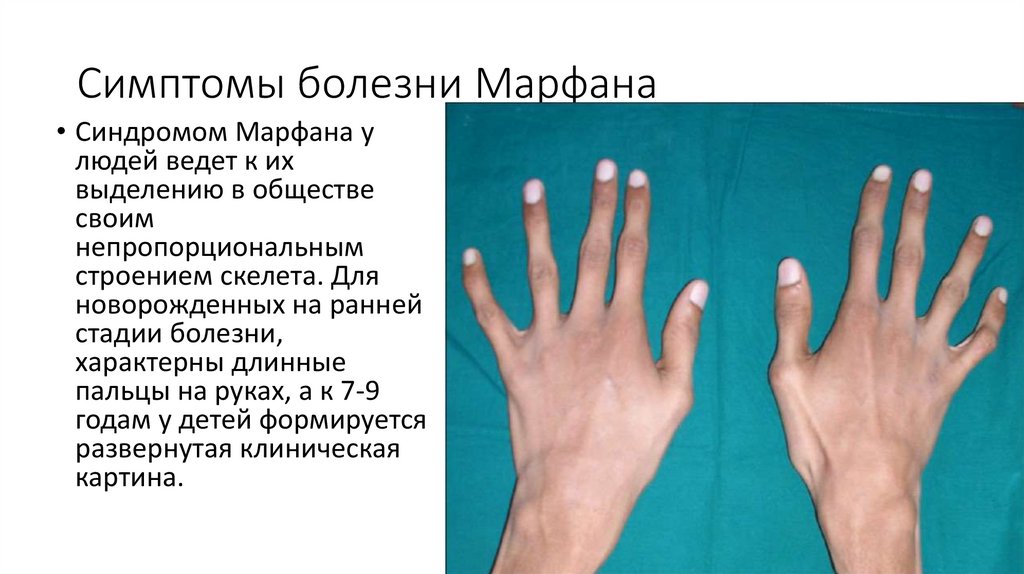
Симптомы болезни Марфана

Синдромом Марфана у людей ведет к их выделению в обществе своим непропорциональным строением скелета. Для новорожденных на ранней стадии болезни, характерны длинные пальцы на руках, а к 7-9 годам у детей формируется развернутая клиническая картина.
У взрослых характерна различная симптоматика в зависимости от системы поражения:
- Нервная система: болезненность в поясничной области, головные боли,поражение симпатической и парасимпатической иннервации органов брюшной полости и малого таза (слабость кишечной стенки, недержание мочевого пузыря у ребенка). Также высок риск развития инсульта, субарахноидального кровоизлияния и разрыва аневризм головного мозга.
- Сердечно – сосудистая система: \пороки сердца (сужение легочной артерии, пролабирование створок двустворчатого клапана, дилатационная кардиомиопатия, расширение границ сердца (аорты и всех ее отделов), дефекты МЖП и МЖЖ перегородок. У больных может развиваться нарушение ритма и проводимости в виде аритмий.
- Опорно-двигательный аппарат: телосложение астенической формы (дети худые), высокий рост у мужчин 190±10 см, у женщин 179±8 см, слаборазвитый подкожно-жировой слой, длинные пальцы (паукообразные), плоскостопие, череп и лицо вытянутые и узкие, недоразвитость скул, нарушение развития зубов и прикуса, вытянутая нижняя челюсть, готическое верхнее небо, (смотрите на картинки выше). С возрастом ребенка может прогрессировать деформация позвоночного столба, с развитием сколиоза. Также может деформироваться грудная клетка, образуется вдавление – «грудь сапожника». Деформированный тазобедренный сустав нередко приводит к инвалидности при неоказанном своевременном лечении.
- Орган зрения: смещение хрусталика за счет слабого связочного аппарата) на ранней стадии, уплощение роговицы, развитие близорукости или дальнозоркости, спазм аккомодации, отслоение сетчатки.
- Кожа и мягкие ткани: перерастяжение кожи с образованием стрий атрофического характера. Они возникают внезапно, не связаны с колебание веса людей, беременностью и гормональным фоном. Кожа липкая, потная, с мраморным оттенком. Подкожно-жировой слой слабо развит, поэтому у больных наблюдаются грыжевые выпячивания в области передней брюшной стенки.
- Дыхательная система: развитие буллезной эмфиземы легких, проявляющейся кашлем, одышкой, развитием дыхательной недостаточности и спонтанного пневмоторакса.
Другие признаки:
- развитие опущения почек (нефроптоз);
- выпадение органов малого таза (опущение матки у женщин, или ее полное выпадение);
- варикоз вен нижних конечностей;
- запоры.
Почему мальчики не болеют
Учитывая, что мутирующий ген несет в себе Х-хромосома, то девочки в плане заболевания находятся в более «выигрышной» позиции. У них присутствует две Х-хромосомы. Поэтому если одна из них «бракованная», то вторая функционирует нормально. Это дает девочке хоть малый шанс на нормальное существование.
У мальчика Х-хромосома одна. Если она имеет мутационный ген, значит, выпадает из работы полностью, и ее нечем заменить. Такие малыши мужского пола, как правило, погибают еще внутриутробно, так и не родившись. Поэтому синдром Ретта у мальчиков встречается крайне редко.
Но, несмотря на такую особенность заболевания, очень редко, но все-таки мальчики с подобным синдромом выживают. Это может быть связано с тем, что не все гены в Х-хромосоме подвергаются мутации. Из-за этого заболевание развивается не столь остро.
Другая причина – наличие у мальчика синдрома Клайнфельтера. При этом наблюдается полисомия половых хромосом, то есть их набор составляет ХХУ. И, если одна Х-хромосома имеет патологический ген, то вторая может регулировать синтез белка и дарить мальчику возможность жизни. Получается такая же картина, как и у девочки.
Лечение синдрома Марфана
От синдрома Марфана полностью избавиться и устранить механизм его развития невозможно. Лечение базируется на улучшении общего состояния больного, устранении клинических проявлений и проведении профилактических мероприятий, препятствующих развитию осложнений.
Больным с данным синдромам рекомендовано ограничить физическую нагрузку до низкого уровня, или минимального. Риск появления патологии сердечно-сосудистой системы возрастает при средних и высоких физических нагрузках.








Следует обходить стороной и повседневные нагрузки, при которых возможно повышение внутригрудного давления, ведущему к развитию пневмоторакса (например, подъем тяжестей, подъем по этажам).
Медикаментозное лечение
Медикаментозная терапия направлена на устранение клинической картины заболевания.
Со стороны сердечно-сосудистой системы рекомендованы β-адреноблокаторы (например: Анаприлин), которые снижают скорость распространения пульсовых волн при стремительно растущем расширении аорты и обратного тока крови на двустворчатом клапане или клапане аорты.
β-адреноблокаторы также оказывают положительный эффект при нарушении ритма и проводимости, в сочетании с сердечными гликозидами.
Но следует помнить о существующих противопоказаниях данных групп препаратов:
- хронический обструктивный бронхит;
- бронхиальная астма;
- снижение частоты сердечных сокращений;
- низкое артериальное давление.
Блокаторы каналов кальция используются при наличии противопоказаний к В-адреноблокаторам.
Хирургия
Хирургическое лечение проводится, если есть осложнения со стороны сердечно-сосудистой системы, с целью коррекции пораженных участков. Его проводят при пролабировании двустворчатого клапана и расслаивании аорты.
При этом осуществляется протезирование двустворчатого клапана.
У беременных с тяжелым течением болезни Марфана, роды разрешаются хирургическим путем.





